一、引言
细胞作为生命的基本单位,承载着遗传信息并执行着复杂多样的生物学功能。然而,细胞之间存在着显著的异质性,这种异质性不仅体现在细胞类型和状态上,还体现在基因表达模式和分子调控机制中。传统的基因表达分析方法,如基于群体细胞的RNA测序(bulk RNA-Seq),虽然能够提供整体的转录组信息,但却掩盖了单个细胞之间的差异。这种“平均化”的数据使得我们难以深入了解细胞间的复杂相互作用以及细胞在不同生理和病理状态下的独特行为。
近年来,单细胞转录组测序(scRNA-Seq)技术的出现和发展,为研究细胞异质性提供了强大的工具。分析单个细胞的转录组,可以揭示不同细胞类型、细胞状态以及细胞间的相互作用,从而更深入地理解细胞在发育、健康和疾病中的作用。然而,单细胞转录组分析面临着诸多挑战,包括细胞内RNA的低丰度、RNA种类的多样性以及转录本长度的不一致性。这些因素使得单细胞转录组的全面和准确分析变得极具挑战性。
幸运的是,随着测序技术的不断进步,尤其是第三代测序技术(TGS)的出现,长读长单细胞转录组测序(long-read scRNA-Seq)逐渐成为可能(见图1)。与传统的第二代测序技术(NGS)相比,TGS技术能够提供更长的读长,从而更好地覆盖完整的转录本,包括复杂的基因融合事件、可变剪接等。这些技术的进步不仅提高了转录组分析的分辨率,还为研究细胞异质性提供了更全面的视角。
本文详细介绍了长读长单细胞转录组测序技术的最新进展,包括其在样本制备、文库构建和生物信息学分析方面的创新,还将探讨这些技术如何帮助我们更好地理解细胞的异质性,并为未来的生物学研究和临床应用提供新的思路和方法。
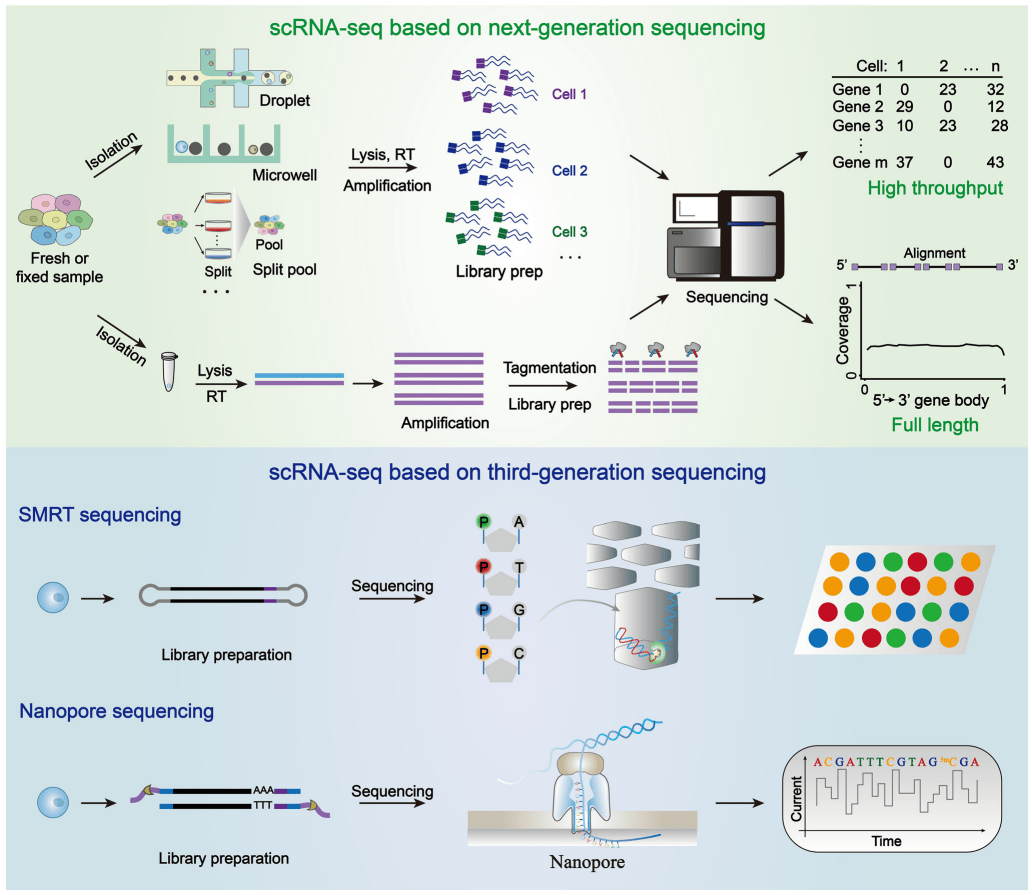 图1
图1
二、基于二代测序的单细胞转录组
自2005年下一代测序(NGS)技术问世以来,其在核酸分析领域引发了革命性的变革。通过在单次运行中同时执行大量测序反应,NGS技术极大地提高了测序通量和速度,同时降低了成本。2009年,Tang等人开发了首个基于NGS的单细胞RNA测序(scRNA-seq)方法,这标志着单细胞转录组学研究的新纪元。此后,各种基于NGS平台的scRNA-seq方法不断涌现,为转录组研究提供了前所未有的视角。
NGS-based scRNA-seq的工作流程主要包括以下步骤:单细胞分离、细胞裂解、逆转录(RT)、扩增、片段化以进行文库制备,以及测序。通过整合细胞条形码策略,众多高通量scRNA-seq技术得以开发,能够在数千个单细胞中同时分析转录组信息。然而,NGS平台生成的短读长(通常为50-150 bp的配对端读)无法获得cDNA🧬的全长信息。为了获得全长读取,通常需要为每个单细胞扩增子进行单独的文库制备,并通过与基因组比对来组装这些基因片段,从而获得全长读取。
jrhz.info本部分将重点关注两类主要的scRNA-seq方法:一是高通量scRNA-seq方法,能够对大量单细胞进行分析;二是全长scRNA-seq方法,用于检测基因剪接、新转录本、基因融合等特征。
2.1 高通量scRNA-seq方法
组织由大量细胞组成,高通量scRNA-seq方法的发展有助于通过同时分析数千个细胞来全面理解组织结构和功能。迄今为止,越来越多的高通量scRNA-seq方法得以开发,大致可以分为两类:基于条形码珠的细胞条形码和基于分裂-池策略的细胞条形码。前者利用单细胞和条形码珠在单独的隔室中配对,通过条形码珠上的寡核苷酸标记mRNA。条形码珠由特定的寡核苷酸组成,包括用于区分不同细胞转录本的细胞条形码、用于消除PCR产物的唯一分子标识符(UMI),以及用于捕获含有poly(A)尾的转录本的oligo(dT)序列。为了实现单细胞与单珠的配对,各种微流控平台,如基于微阀、液滴和微孔的平台,已被开发用于高通量scRNA-seq。后者则利用分裂-池策略为每个细胞分配独特的组合条形码,通过高容量条形码组合实现超高通量单细胞的条形码标记。然而,由于单细胞与条形码珠配对效率低或因多轮分裂和混合导致细胞丢失,细胞利用率(即最终通过细胞条形码识别的单细胞数量与所有输入细胞的比率)较低。此外,固定样本(如临床存档的固定样本和因无法立即处理而必须固定的样本)也越来越受到关注。为了实现固定样本的分析并解决相关问题,开发了更灵活和灵敏的scRNA-seq方法。本节将简要回顾先进的高通量scRNA-seq方法,重点关注提高细胞利用率和扩大样本适用性,特别是针对固定样本。
2.1.1 高通量单细胞转录组测序技术的改进:提高细胞利用率
在单细胞转录组测序(scRNA-seq)领域,细胞利用率是一个关键指标,它直接影响到实验的成功率和数据质量。细胞利用率指的是最终通过细胞条形码识别的单细胞数量与所有输入细胞的比率。传统的高通量scRNA-seq方法在细胞利用率方面存在瓶颈,尤其是在处理低输入样本时,细胞丢失问题尤为突出。为了提高细胞利用率,近年来研究者们开发了一系列先进的高通量scRNA-seq方法,这些方法通过优化微流控平台和液滴微流控技术,显著提高了单细胞与条形码珠的配对效率。
微流控平台的创新
微流控技术因其微型化、集成化和并行操作的优势,在高通量单细胞转录组测序中占据主导地位。然而,传统的微流控平台在细胞利用率上仍受限于泊松分布。为了提高细胞利用率,研究者们开发了多种创新策略。
例如,Fan团队提出了dTNT-seq技术,通过整合介电电泳(DEP)和转移策略,实现了单细胞与条形码珠的高效配对。具体而言,单细胞首先被捕获在20微米的纳米孔阵列上,随后通过将『芯片』翻转,将细胞转移到更大的50微米微孔中进行后续的条形码珠捕获。这种设计实现了双亚泊松分布,单细胞与珠子的配对率达到了约75%(见图2)。
 图2
图2
进一步改进的Well-paired-seq技术则采用了双层微孔平台,利用尺寸排阻原理将单细胞和单珠分别捕获到底部和顶部微孔中。该方法不仅允许单细胞的累积捕获,还能有效进行缓冲液交换和去除无细胞RNA,显著提高了配对效率(约82%)和数据质量(见图2b)。
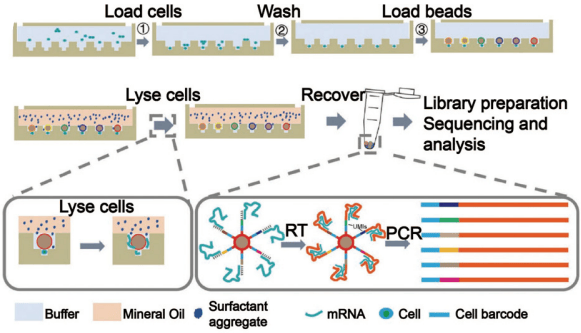 图3
图3
液滴微流控技术的发展
液滴微流控技术因其最小化污染、高通量以及不受细胞大小限制等优点,成为高通量单细胞转录组测序的另一重要平台。然而,传统的液滴微流控技术在细胞利用率和低输入样本处理方面仍存在不足。
Bues等人开发的Disco系统通过结合机器视觉和微阀操作,实现了高捕获和配对效率。Disco『芯片』利用Quake风格的微阀主动检测细胞,实现了超过91.4%的液滴中包含一个细胞和一个珠子的高配对效率(见图4)。此外,InDrop技术通过在液滴中加载软且可变形的水凝胶珠子,实现了接近100%的液滴占用率和高单细胞/单珠子配对效率(约90%)。
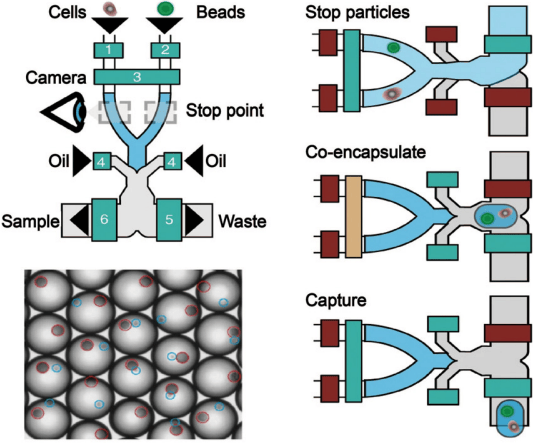 图4
图4
分裂-池策略的优化
分裂-池策略通过多轮分裂-池循环为单细胞转录本分配组合条形码,实现了超高通量单细胞转录组分析。Cao等人开发的sci-RNA-seq是首个基于分裂-池的scRNA-seq方法,通过在固定和通透化细胞后进行原位逆转录(RT)反应,引入第一轮条形码,随后通过荧光激活细胞分选(FACS)将细胞重新分配到微孔板中进行第二轮条形码引入(见图5)。
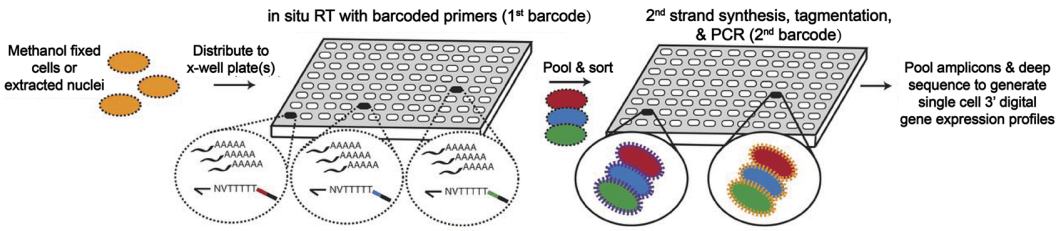 图5
图5
为了减少繁琐的分裂-池操作,研究者们进一步将分裂-池策略与液滴/微孔平台相结合。例如,scifi-RNA-seq和Microwell-seq 2.0分别利用液滴和微孔平台进行第二轮条形码引入(见图3b)。这些方法不仅提高了细胞利用率,还通过多轮条形码组合克服了泊松分布的限制。
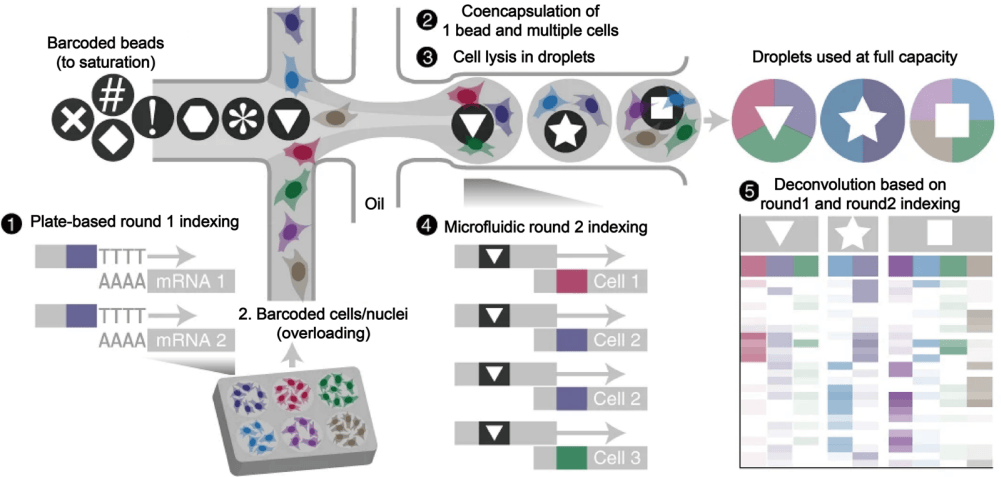 图6
图6
5′端单细胞转录组测序技术的突破
与3′端scRNA-seq方法相比,5′端scRNA-seq方法在免疫受体库分析和揭示顺式调控元件方面具有优势,但其通量较低。为了克服这一限制,Li等人开发了FIPRESCI技术,通过组合索引实现了高通量5′端单细胞转录组测序。该方法在固定、通透化和原位RT后,利用Tn5转座酶引入微孔特异性条形码,随后通过液滴特异性条形码对5′端转录本进行标记。FIPRESCI技术能够从约10万个单细胞中进行高通量单细胞转录组分析,比标准10× Genomics 5′ scRNA-seq方法的通量提高了约10倍(见图7)。
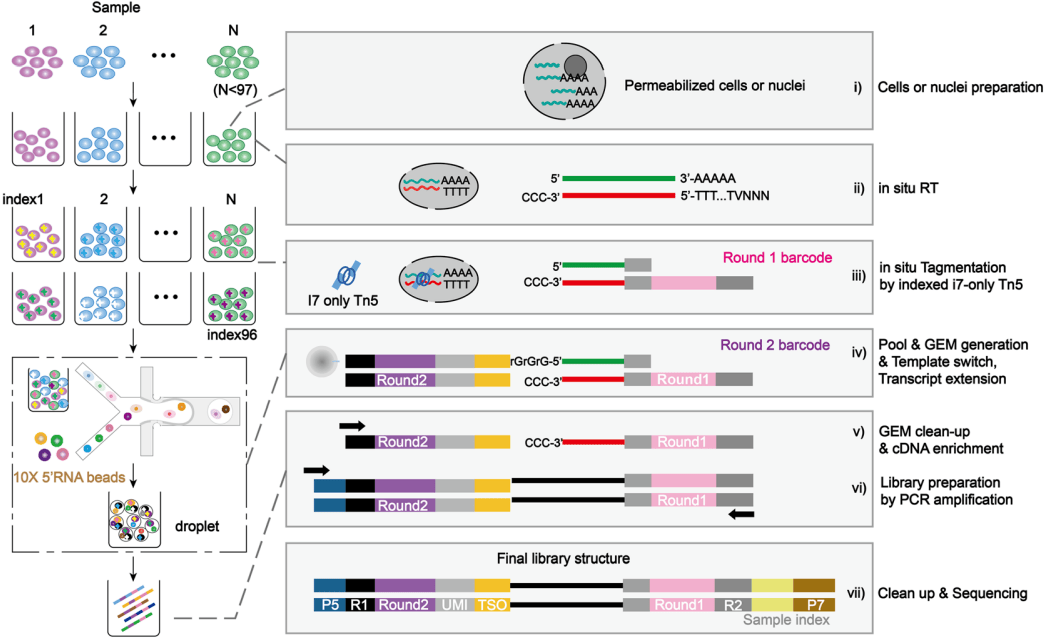 图7
图7
2.1.2 高通量单细胞转录组测序技术的改进:扩展样本适用性
在单细胞转录组测序(scRNA-seq)的研究中,样本的多样性是一个重要的考量因素。传统的高通量scRNA-seq方法主要适用于新鲜组织或培养细胞,而对于固定样本(如福尔马林固定石蜡包埋FFPE样本)的支持有限。然而,固定样本在临床诊断和疾病研究中具有重要的应用价值,因为它们是临床存档中最丰富的样本类型。因此,开发能够处理固定样本的scRNA-seq方法,对于扩展样本适用性和提高研究的临床相关性至关重要。
酒精固定样本的处理
酒精类固定剂(如甲醇)通过脱水作用固定细胞和组织,不会导致核酸的化学交联,因此在固定后可以通过再水化恢复核酸的完整性,对RNA质量的影响较小。Alles等人首次将基于甲醇固定的高通量scRNA-seq方法应用于Drop-seq平台(见图8),并成功地在多种细胞类型(包括果蝇胚胎、小鼠大脑和神经细胞)中实现了高质量的转录组分析。然而,甲醇固定在处理某些富含RNase和蛋白酶的原代细胞类型(如免疫组织)时效果不佳。此外,甲醇固定可能导致RNA泄漏,增加背景噪声。
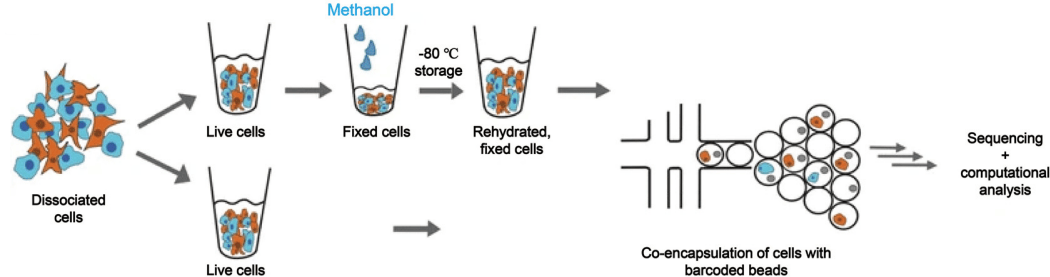 图8
图8
醛类固定样本的处理
与甲醇不同,醛类固定剂(如甲醛、PFA和福尔马林)通过形成蛋白质和核酸之间的共价化学键来固定样本,从而减少RNA泄漏的风险。Phan等人开发了FD-seq技术,用于PFA固定细胞亚群的分析。在FD-seq中,通过在液滴内加热至56°C来逆转PFA交联,从而实现RNA的完全释放,并通过在裂解缓冲液中加入蛋白酶K来提高RNA产量。FD-seq能够检测到比甲醇固定方法更多的基因和转录本。
单细胞核转录组测序(snRNA-seq)技术的发展
对于一些脆弱或敏感的细胞类型,单细胞转录组测序可能在组织分离过程中导致细胞丢失或基因表达变化。单细胞核转录组测序(snRNA-seq)通过快速获取单个细胞核来解决这一问题。尽管细胞核中的RNA量低于整个细胞,但snRNA-seq仍能提供与单细胞转录组测序相当的基因检测和细胞类型识别能力。然而,现有的大多数snRNA-seq方法主要关注具有poly(A)尾的蛋白编码RNA。近年来,越来越多的研究表明,非编码RNA(如miRNA、lncRNA和环状RNA)在细胞命运和功能调控中发挥重要作用。
为了克服这一限制,Niu等人开发了MATQ-Drop技术,用于小鼠和人类大脑的单突触和细胞核的全转录组测序(见图9)。在3% PFA固定和细胞核通透化后,通过多轮退火反应,使引物能够高效地与所有转录本的内部区域杂交,从而实现高效的全转录组捕获。与10× Genomics v3.1平台相比,MATQ-Drop的基因检测灵敏度提高了135%。
 图9
图9
FFPE样本的处理
FFPE样本广泛用于临床样本保存,为疾病诊断和治疗提供了宝贵的资源。然而,从FFPE样本中分离完整的单细胞和捕获RNA仍然具有挑战性,因为FFPE样本中的RNA可能发生交联、修饰和降解。为了克服这些限制,Xu等人开发了snRandom-seq技术,用于FFPE样本的单细胞核全转录组测序(见图10)。通过从FFPE样本中获取完整的细胞核,进行分离和通透化处理,然后使用带有预标记的oligo(dT)和随机引物进行原位逆转录,从而实现单细胞核的全转录组测序。
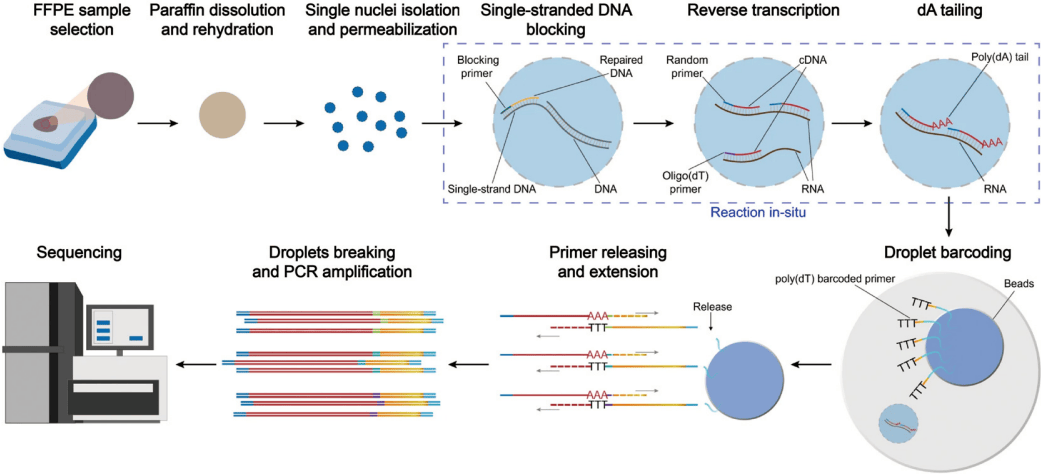 图10
图10
2.2 全长单细胞转录组测序方法(Full-length scRNA-seq methodologies)
全长单细胞转录组测序(full-length scRNA-seq)方法能够提供单个细胞转录组的完整信息,这对于理解基因表达调控、RNA剪接和转录本异构体的多样性至关重要。与传统的短读长单细胞转录组测序方法相比,全长scRNA-seq能够覆盖完整的转录本,从而更准确地检测基因融合、可变剪接事件以及转录本的全长结构。近年来,研究者们开发了多种全长scRNA-seq方法,这些方法在灵敏度、准确性和通量方面各有优势。
早期全长scRNA-seq方法
Tang等人在2009年首次报道了能够对单细胞全长转录本进行测序的技术(见图11)。该方法通过使用oligo(dT)引物捕获mRNA,然后在第一链cDNA🧬上添加poly(A)尾,接着利用另一对oligo(dT)引物合成第二链cDNA🧬。通过增加逆转录(RT)和PCR延伸时间,可以生成长达3kb的cDNA🧬。然而,这种方法无法检测非poly(A)尾的RNA,这些RNA在基因表达调控和细胞功能决定中起着关键作用。
 图11
图11
为了同时检测poly(A)+和poly(A)− RNA,Fan等人开发了单细胞通用poly(A)独立RNA测序(SUPeR-seq)方法(见图12)。该方法使用随机引物而不是oligo(dT)引物进行cDNA🧬合成,从而能够检测到包括环状RNA在内的多种非poly(A) RNA。通过SUPeR-seq,研究者们从单个HEK293T细胞和小鼠早期胚胎中分别鉴定出141和2891个环状RNA转录本,以及一系列poly(A)− RNA,这有助于揭示poly(A)− RNA的功能和调控机制。
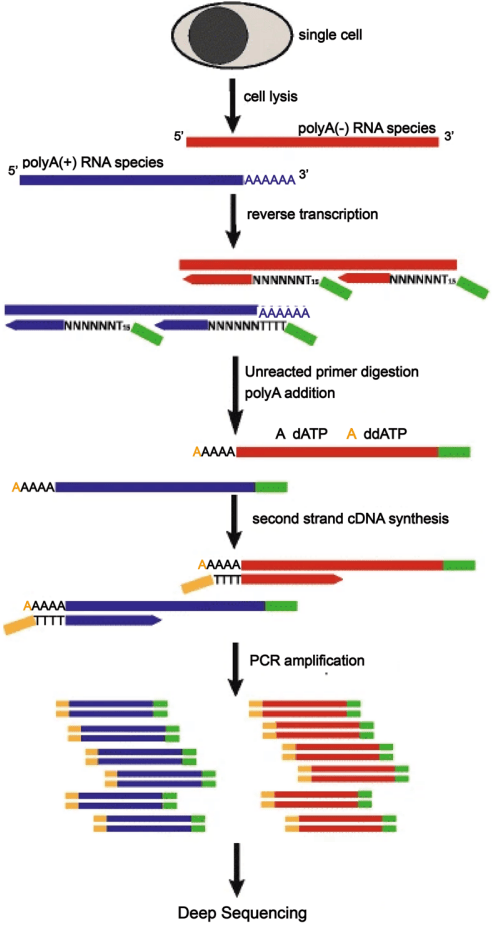 图12
图12
模板转换策略的改进
尽管poly(A)尾方法能够提供全长转录本信息,但其操作繁琐,且由于3′端偏倚导致覆盖不均匀。为了改善覆盖范围,Smart-seq方法利用MMLV逆转录酶的模板转换活性,而不是poly(A)尾,来合成第二链cDNA🧬。利用MMLV逆转录酶的模板转换和末端转移酶活性,可以在第一链cDNA🧬的3′端添加几个未模板化的胞嘧啶,从而合成第二链cDNA🧬(见图13)。Smart-seq显著提高了转录组覆盖范围,简化了操作流程,有助于鉴定可变剪接事件。然而,Smart-seq在RNA异构体定量方面的准确性仍有待提高。
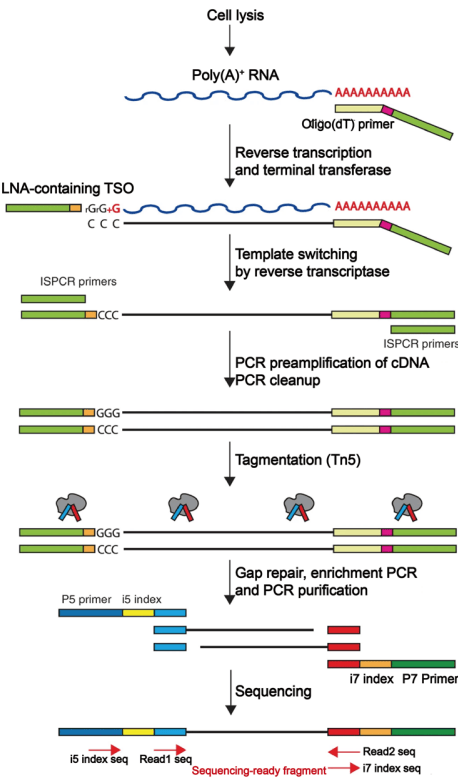 图13
图13
为了进一步提高灵敏度和准确性,Hagemann-Jensen等人开发了Smart-seq3(见图14)。该方法通过在特殊模板转换引物(TSO)中加入含有UMI序列和Tn5基序的混合物,在测序过程中生成可区分的内部读取和5′端UMI包含的读取。此外,通过计算机辅助重建具有相同UMI的不同cDNA🧬分子的片段,可以鉴定等位基因来源和异构体。Smart-seq3的改进灵敏度有助于更准确地区分细胞簇和状态,揭示不同组织中的异构体模式。
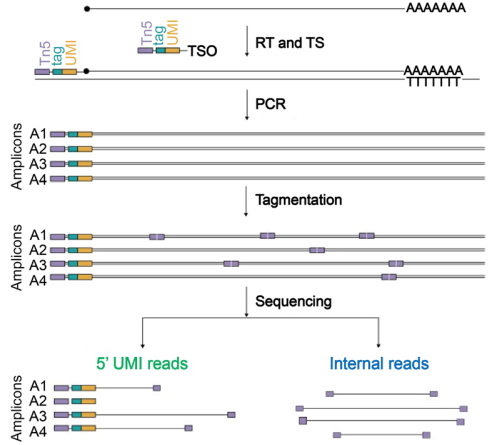 图14
图14
三、基于三代测序的单细胞转录组(TGS-based long-read scRNA-seq)
随着单细胞转录组测序技术的不断发展,研究者们对更高分辨率和更全面的转录组分析的需求日益增加。传统的二代测序(NGS)技术虽然在高通量和准确性方面表现出色,但由于其短读长的限制,在分析复杂的转录本结构(如基因融合、可变剪接和RNA修饰)时存在不足。为了克服这些限制,三代测序技术(TGS),特别是单分子实时测序(SMRT)和纳米孔(nanopore)测序,逐渐成为单细胞转录组测序的新选择。
三代测序技术(TGS)以其长读长和单分子实时测序能力而闻名,能够直接对超过10 kb的原生DNA🧬进行测序,并检测结构变异、大型基因组重排和相位单倍型。与二代测序(NGS)平台相比,TGS技术能够提供更完整的转录本信息,从而更准确地分析基因表达模式和转录本异构体。近年来,基于TGS的单细胞转录组测序方法取得了显著进展,这些方法不仅提高了转录组分析的分辨率,还为研究细胞异质性提供了更全面的视角。
本部分将详细介绍基于TGS的单细胞转录组测序技术的最新进展,包括其在样本制备、文库构建和生物信息学分析方面的创新,还会探讨这些技术如何帮助更好地理解细胞的异质性,并为未来的生物学研究和临床应用提供新的思路和方法。
3.1 基于 PacBio SMRT 测序的长读长单细胞转录组测序
PacBio 单分子实时(SMRT)测序技术以其长读长和单分子实时测序能力而闻名,能够直接对超过 10 kb 的原生 DNA🧬 进行测序,并检测结构变异、大型基因组重排和相位单倍型。这种技术能够生成超过 1 kb 的长读长,从而更准确地覆盖完整的转录本,包括复杂的基因融合事件和可变剪接。近年来,研究者们开发了多种基于 PacBio SMRT 测序的单细胞转录组测序方法,这些方法在提高转录组分析的分辨率和准确性方面表现出色。
ScISOr-Seq 方法
Gupta 等人开发了单细胞异构体 RNA 测序(ScISOr-Seq)方法,该方法结合了 NGS 和 TGS 平台的优势(见图 15)。通过在 10× Genomics Chromium 平台上对单细胞进行条形码标记,ScISOr-Seq 能够在保持细胞来源的同时,利用 PacBio 长读长测序技术检测 RNA 异构体。这种方法不仅能够检测已知和新的异构体,还能通过链接短读和长读测序数据,将 RNA 异构体表达分配到单个细胞中。ScISOr-Seq 在分析小鼠小脑的 1000 多个单细胞时,成功检测到了大量的异构体,显著提高了对 RNA 结构的理解和基因组注释的准确性。
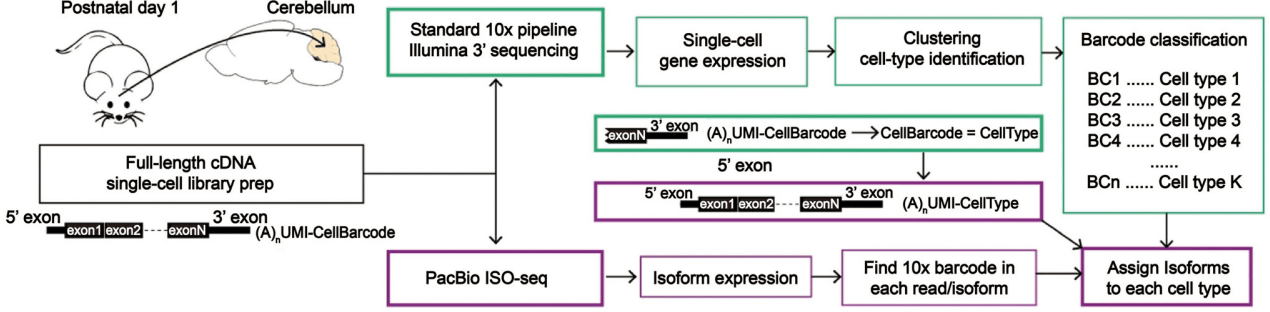 图15
图15
CCS 系统
为了提高测序的准确性,PacBio 开发了circulating consensus sequencing(CCS)系统(见图16)。CCS 系统通过将线性 DNA🧬 分子的两端连接发夹适配器,生成闭合环状的 SMRTbell 模板。通过多次读取每个 SMRTbell 模板,可以获得多个重叠的共识序列。这种技术不仅提高了测序的准确性(高达 99.8%),还能生成平均长度为 13.5 kb 的长读长。然而,CCS 系统的通量较低,成本较高,限制了其在大规模单细胞转录组分析中的应用。
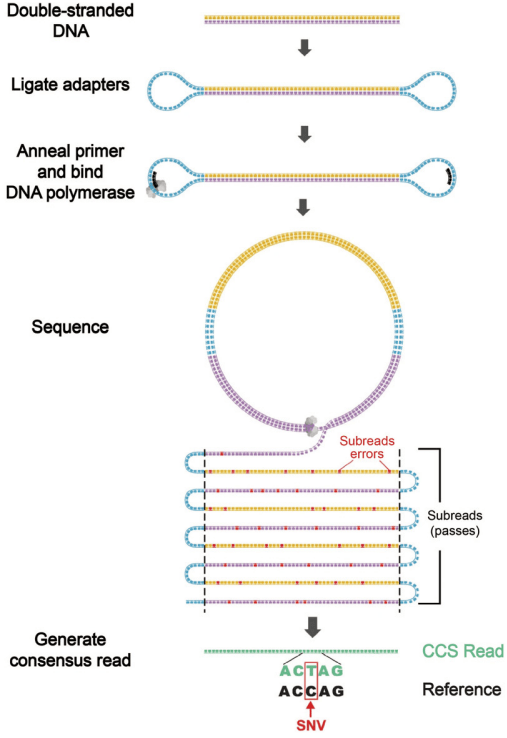 图16
图16
HIT-scISOseq 方法
为了克服 CCS 系统的通量限制,Zheng 等人开发了高通量单细胞异构体测序(HIT-scISOseq)方法(见图 17)。该方法首先通过 10× Genomics 生成细胞特异性的全长 cDNA🧬 序列,然后通过生物素标记的 PCR 引物富集单细胞全长 cDNA🧬 产物,并消除 TSO 伪影。通过将多个 cDNA🧬 连接成头尾相连的长插入 SMRTbell 模板进行测序,HIT-scISOseq 能够利用 CCS 技术的全部长读长测序潜力。这种方法不仅提高了数据输出量(比标准 scISOseq 方法高出 8 倍),还能通过细胞条形码区分每个 cDNA🧬。
 图17
图17
MAS-ISO-seq 方法
为了最大化 PacBio 平台的测序通量和能力利用率,Al'Khafaji 等人开发了 MAS-ISO-seq 方法,用于将 DNA🧬 片段编程连接成长测序模板(见图 18)。通过去除 TSO 伪影,并使用链霉亲和素/生物素选择法,MAS-ISO-seq 能够将 cDNA🧬 库分成多个池进行独立 PCR,加入池特异性的 dU 含条形码适配器。随后,将所有扩增的 cDNA🧬 库合并进行 dU 消化,这些带有粘性末端的 cDNA🧬 通过条形码引导的连接反应连接起来。MAS-ISO-seq 不仅将通量提高了 15 倍以上,还能实现每轮运行近 4000 万 cDNA🧬 读长,极大地提高了 PacBio 平台的利用率。
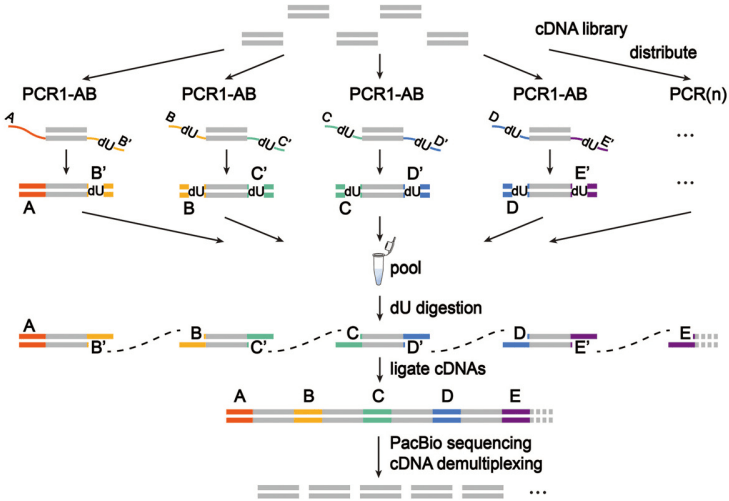 图18
图18
3.2 基于 Oxford Nanopore 测序的长读长单细胞转录组测序
Oxford Nanopore 测序技术以其长读长、低成本和便携性而闻名。该技术通过测量单个 DNA🧬 或 RNA 分子通过纳米孔时的电流变化来实现测序。与 PacBio SMRT 测序相比,Oxford Nanopore 测序不需要 DNA🧬 扩增,从而避免了扩增偏差和在高 GC 区域的测序问题。然而,Oxford Nanopore 测序的错误率限制了其在大规模单细胞转录组分析中的应用。近年来,研究者们开发了多种基于 Oxford Nanopore 测序的单细胞转录组测序方法,这些方法在提高测序准确性和通量方面取得了显著进展。
SCAN-seq 方法
Fan 等人开发了单细胞全长 RNA 扩增和测序(SCAN-seq)方法,利用 Oxford Nanopore 平台进行单细胞转录组测序(见图 19)。该方法通过使用带有条形码的逆转录引物对单细胞 RNA 进行逆转录和扩增,然后将多个单细胞的 cDNA🧬 合并进行纳米孔文库构建和测序。SCAN-seq 能够检测到小鼠胚胎发育过程中 27,250 个未注释的转录本,并确定单细胞内的等位基因特异性基因表达模式。然而,由于缺乏唯一分子标识符(UMI)以消除 PCR 扩增错误,该方法在基因表达分析中存在偏差。
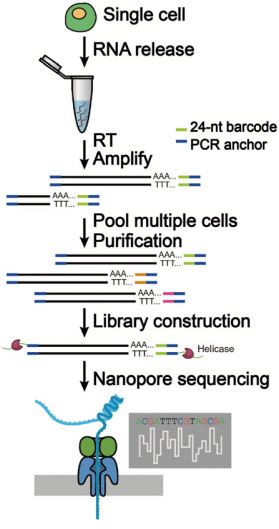 图19
图19
ScNaUmi-seq 方法
为了克服这一限制,Lebrigand 等人开发了单细胞纳米孔测序与 UMI(ScNaUmi-seq)方法,通过结合长读长纳米孔测序和短读长测序来实现高准确性条形码分配(见图 20)。通过在 10× Genomics 平台上生成带有条形码的 cDNA🧬,然后分别在 Illumina 平台和纳米孔测序平台上进行文库制备和测序,ScNaUmi-seq 能够通过 Illumina 数据的高准确性来指导纳米孔读取的条形码分配。这种方法不仅提高了细胞条形码和 UMI 分配的准确性(分别为 99.8% 和 97.4%),还能通过比较 Illumina 和纳米孔数据来解决测序数据中的错误。
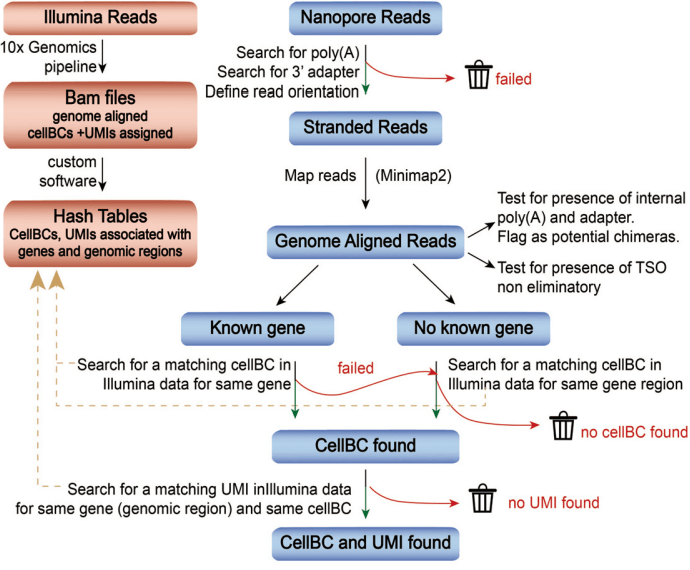 图20
图20
RAGE-seq 方法
为了分析 T 细胞和 B 细胞的抗原受体多样性,Singh 等人开发了抗原受体和基因表达测序(RAGE-seq)方法,该方法结合了高通量单细胞转录组测序和长读长测序(见图 21)。通过在液滴微流控平台上生成带有条形码的全长 cDNA🧬 库,然后将 cDNA🧬 分成两部分:一部分用于短读长测序以测量 3′ 端基因表达并生成高准确性的细胞条形码序列,另一部分用于靶向捕获 B 细胞和 T 细胞受体转录本。通过长读长 Oxford Nanopore 测序,RAGE-seq 能够将单个细胞的转录组谱和全长抗原受体序列链接起来,从而揭示不同克隆型的基因表达特征和特定克隆型的分化状态。
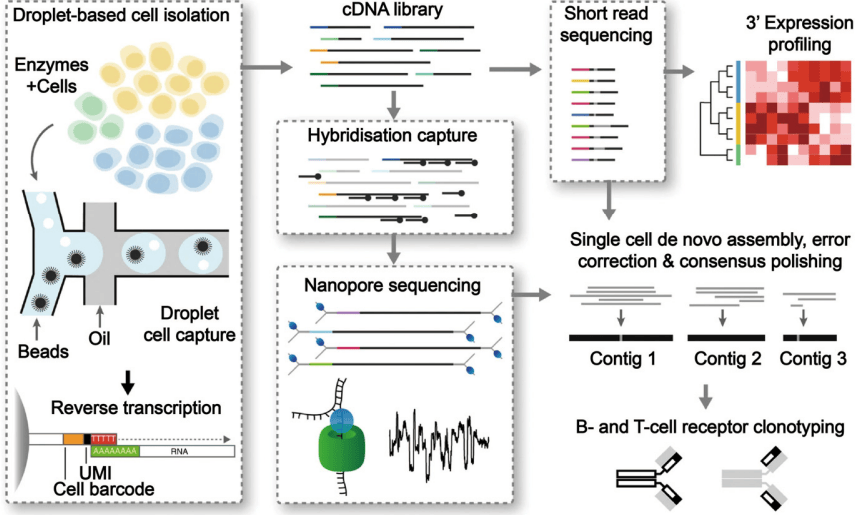 图21
图21
SCAN-seq2 方法
为了进一步提高测序的准确性和通量,Liao 等人开发了 SCAN-seq2 方法,用于高通量和高灵敏度的单细胞全长转录组测序(见图 22)。该方法通过在 96 孔板中进行逆转录时引入 3′ 端条形码和 UMI,然后将 N 个不同的条形码单细胞合并到 M 个管中进行 PCR 扩增,最后在纳米孔测序平台上同时测序 N×M 个单细胞。SCAN-seq2 不仅提高了测序的准确性(由于引入了 UMI),还降低了成本(比 SCAN-seq 低约 20 倍),并且在获得每个转录本全长片段的覆盖度时所需的测序深度比短读长测序更低。
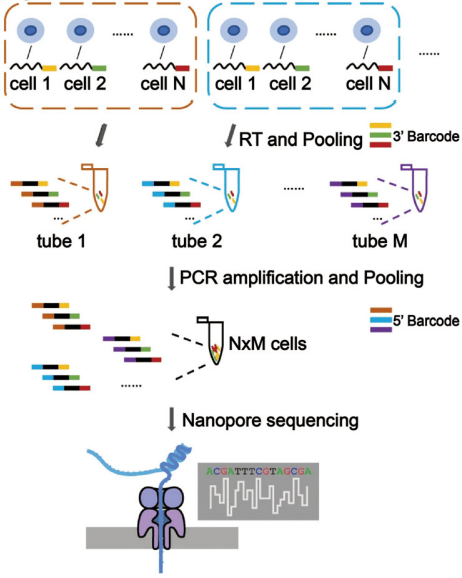 图22
图22
四、结论与展望
本综述全面总结了单细胞转录组测序(scRNA-seq)技术的进展,特别是基于二代测序(NGS)和三代测序(TGS)的高通量和长读长单细胞转录组测序方法。这些技术的发展极大地推动了对细胞异质性的理解,为研究细胞功能、基因表达调控以及疾病机制提供了强大的工具。然而,尽管取得了显著进展,单细胞转录组测序领域仍面临一些挑战,同时也展现出广阔的发展前景。
已取得的进展
1. 高通量与长读长的结合:NGS 技术以其高通量和低成本的优势,成为单细胞转录组测序的主流方法。然而,其短读长限制了对复杂转录本结构的分析。TGS 技术,尤其是 PacBio 和 Oxford Nanopore 测序平台,以其长读长和单分子实时测序能力,弥补了 NGS 的不足,能够更准确地检测基因融合、可变剪接和 RNA 修饰等复杂事件。
2. 技术优化与创新:近年来,研究者们开发了多种创新方法,如 ScISOr-Seq、CCS 系统、HIT-scISOseq 和 MAS-ISO-seq 等,这些方法不仅提高了测序的准确性和通量,还通过结合 NGS 和 TGS 平台的优势,实现了更全面的转录组分析。
3. 样本适用性的扩展:为了满足不同研究需求,特别是临床样本的应用,研究者们开发了适用于固定样本(如 FFPE 样本)的 scRNA-seq 方法,如 MATQ-Drop 和 snRandom-seq,这些方法显著扩展了单细胞转录组测序的样本适用性。
面临的挑战
1. 样本制备与 RNA 完整性:尽管技术不断进步,但单细胞转录组测序仍受限于样本制备过程中的 RNA 完整性。固定样本和低输入样本的处理仍然是一个挑战,需要进一步优化 RNA 提取和保护方法。
2. 测序准确性和通量的平衡:TGS 技术虽然提供了长读长,但其错误率和较低的通量限制了大规模应用。如何在提高测序准确性的同时保持高通量,是当前需要解决的关键问题。
3. 多组学整合:单细胞转录组测序提供了丰富的基因表达信息,但细胞功能和状态的调控涉及多个分子层面。如何整合转录组、蛋白质组、代谢组等多组学数据,以全面解析细胞的分子调控网络,是未来研究的重要方向。
4. 计算方法的改进:随着单细胞测序数据量的激增,如何高效、准确地处理和分析这些数据成为一个瓶颈。需要开发更快速、更准确的计算方法,以应对数据的复杂性和高维度。
未来展望
1. 技术融合与创新:未来,单细胞转录组测序技术将朝着更高通量、更长读长和更高准确性的方向发展。结合 NGS 和 TGS 平台的优势,开发新的测序方法和文库制备策略,将是提高单细胞转录组测序性能的关键。
2. 多组学整合分析:通过整合单细胞转录组、蛋白质组、代谢组等多组学数据,能够更全面地解析细胞的分子调控网络,揭示细胞功能和状态的动态变化。这将为理解复杂生物系统和疾病机制提供更深入的见解。
3. 临床应用的拓展:随着技术的成熟和成本的降低,单细胞转录组测序有望在临床诊断、疾病监测和个性化治疗中得到更广泛的应用。特别是对于 FFPE 样本和稀有细胞类型的研究,将为临床医学带来新的突破。
4. 计算方法的发展:随着人工智能和机器学习技术的快速发展,开发更高效、更准确的计算方法,以处理和分析单细胞测序数据,将是未来研究的重要方向。这将有助于从海量数据中提取有价值的信息,加速生物学和医学研究的进展。
总之,单细胞转录组测序技术的发展为生命科学和医学研究带来了新的机遇和挑战。通过不断的技术创新和多学科的交叉合作,我们期待这些技术能够在更广泛的领域中得到应用,为揭示生命奥秘和改善人类健康做出重要贡献。
参考文献
Huang, S., Shi, W., Li, S., Fan, Q., Yang, C., Cao, J., & Wu, L. (2024). Advanced sequencing-based high-throughput and long-read single-cell transcriptome analysis. Lab on a Chip, 24, 2601–2621. https://doi.org/10.1039/d4lc00105b




